Clinevo and Clinion’s Strategic Alliance for an Integrated eClinical Experience
June 21, 2024 | Uncategorized | No Comments

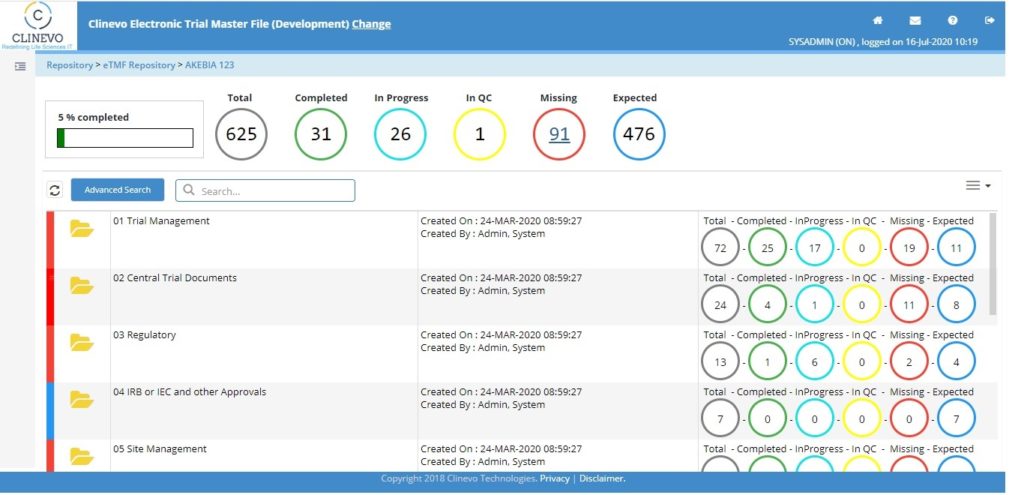
Clinevo, an industry leader in eClinical Safety Solutions, today announced a Strategic Partnership with Clinion, a leading eClinical solutions provider. This collaboration brings together Clinevo’s eTMF and Safety Solutions with Clinion’s AI-enabled eClinical platform, offering a fully integrated seamless end-to-end eClinical Solution.
Through this collaboration, Clinion now offers Clinevo’s eTMF and Safety Solutions alongside our existing suite of products. Similarly, Clinevo will now be able to offer Clinion’s comprehensive EDC, RTSM, CTMS, ePRO, and eConsent solutions to their customers.
Customers would now be able to access both Clinevo and Clinion products on one platform, improving data quality and reducing integration challenges and trial timelines.
Clinion’s robust suite of eClinical tools, making them an ideal complement to Clinevo’s eTMF and Safety Solutions designed to enhance regulatory compliance and streamline safety reporting. This partnership reflects a shared commitment to innovation and excellence in clinical trial management.
“We are delighted to join forces with Clinion to provide AI enabled end-to-end Integrated Clinical Solutions to our customers. Their innovative EDC, CTMS, RTSM, and ePRO solutions are a perfect match for our eTMF and Safety offerings. Together, we can provide a seamless and integrated eClinical experience that supports our clients in conducting efficient and compliant clinical trials.”
– Arunkumar Devaraj, Director of Clinevo
“We are excited to partner with Clinevo to expand our portfolio and deliver even greater value to our customers, By combining our strengths, we can offer a holistic eClinical experience that streamlines clinical trials, improves data accuracy, and accelerates the development of new therapies.”
– Manuj Vangipurapu, CEO of Clinion
This strategic partnership represents a significant step forward for both Clinion and Clinevo, positioning them as leaders in the eClinical solutions market. By leveraging each other’s strengths, the companies aim to enhance the efficiency and effectiveness of clinical trials, ultimately benefiting sponsors, CRO’s, and patients alike.
About Clinevo
Clinevo is a prominent provider of drug safety and eTMF solutions. Clinevo offers end-to-end drug safety solutions, including the PV database, AS2 gateway, literature automation platform, signal detection platform, MICC, web, and email intake solutions for drug safety and pharmacovigilance.
About Clinion
Clinion is a global clinical technology company that offers AI-enabled eClinical solutions consisting of EDC, RTSM, CTMS, eCOA and Document Automation that cover the entire clinical trial lifecycle. Clinion is committed to innovating the future of clinical trials through AI/ML and empowering its partners to manage trials more efficiently at lesser costs.